¿Qué es?
La Miopatía Nemalínica es una enfermedad neuromuscular congénita rara, relativamente no progresiva y caracterizada por debilidad muscular de predominio en la musculatura facial, cervical y de las extremidades, así como por la presencia de unas estructuras en el músculo esquelético llamadas cuerpos nemalínicos.
¿Por qué es rara?
La MN es una Enfermedad Rara pues se estima que afecta a 1 de cada 50.000 nacidos a nivel mundial, estando registrados alrededor de 30 casos en España. Pese a ello es una de las miopatías congénitas más prevalentes, representando un 20% de todas ellas.
¿Qué la caracteriza?
La presentación clínica más habitual se caracteriza por un inicio en la infancia temprana o niñez con:
- Hipotonía (disminución del tono muscular).
- Hiporreflexia (presencia de reflejos clinicomusculares apagados).
- Debilidad generalizada.
- Atrofia muscular (desgaste o pérdida del tejido muscular).
De predominio en músculos faciales, axiales (músculos de abdomen, tórax, dorso, lumbares, cuello y cabeza) y extremidades (superiores e inferiores). Algunas características adicionales son:
- Deformidades esqueléticas.
- Facies dismórficas (rasgos faciales anómalos por debilidad).
- Paladar alto arqueado.
- Distrés respiratorio (insuficiencia respiratoria grave) con infecciones de vías respiratorias.
En conjunto provoca dificultades:
- Respiratorias.
- Deglutorias (disfagia).
- Del habla.
- Motoras (limitaciones posturales, de desplazamiento y coordinación de movimientos).
- Esqueléticas (hipermobilidad articular, contracturas y escoliosis)
La MN presenta además una variabilidad importante tanto en la edad de inicio como en el grado de afectación.
¿Cómo se clasifica?
Pese a la importante variabilidad que la MN presenta tanto en la edad de inicio como en el grado de afectación, se clasifica en:
- Típica congénita: Presenta los primeros síntomas al nacer o en el primer año de vida, siendo estos hipotonía, debilidad y problemas en la alimentación. La afectación respiratoria se debe sospechar si se producen infecciones respiratorias recurrentes o se detecta hipoventilación durante el sueño, siendo la afectación cardíaca rara. En ocasiones los síntomas se inician después del primer año con retraso en el desarrollo motor, marcha miopática (balanceante) y/o problemas de deglución y habla nasal. La debilidad muscular suele ser proximal (muslos), pudiendo ser también distal (pie caído). Su evolución es muy lentamente progresiva e incluso estable.
- Neonatal grave: implica una mayor afectación, presentándose al nacer con hipotonía severa, insuficiencia respiratoria y deglutoria. Requiriendo soporte ventilatorio y alimentación por gastrostomía. El corazón puede verse afectado.
- Amish: solo afecta a dicha población. De aparición neonatal, la expectativa de vida raramente excede los 2 años por complicaciones de tipo respiratorio.
- De inicio tardío: de aparición en la edad adulta y genéticamente determinada.
- Adquirida en edad adulta (Miopatía Nemalínica esporádica de inicio tardío; SLONM): progresiva, genéticamente no determinada y con una prevalencia de 1/1.000.000.
¿Cuál es su origen?
El origen de la MN es genético, encontrándose a día de hoy identificados trece genes como causantes, además recientemente se han identificado dos más (GYG1 y RYR3) que pueden ser potencialmente causantes pero aún están pendientes de estudio.
Dichos genes son:
- ACTA1: causan del 20% al 25% de todas las MN y hasta el 50% de los casos más severos. Participa en la contracción muscular.
- NEB: causan alrededor del 50% de las MN. Participa en la contracción muscular.
- TPM3: deben tenerse en cuenta cuando los cuerpos nemalínicos se localizan exclusivamente en las fibras lentas de tipo 1 o cuando solo se detecta desproporción de tipo de fibras en la biopsia muscular. Participa en la contracción muscular.
- TPM2: deben considerarse sobre todo en las formas leves. Participa en la contracción muscular.
- TNNT1: se han descrito casi exclusivamente en la población Amish. Participa en la contracción muscular.
- CFL2: se ha descrito en muy pocos casos hasta el momento. Participa en la contracción muscular.
- KBTBD13: se han asociado a la presencia de movimientos lentos peculiares sin afectación facial ni respiratoria. Regula sistemas de degradación y remplazo proteico.
- KHLH40: se han identificado sobre todo en pacientes con formas de presentación neonatal severa, contracturas congénitas, fallo respiratorio y dificultades en la deglución. Regula sistemas de degradación y remplazo proteico.
- KHL41: pueden causar distintas formas, desde las más severas neonatales hasta casos de presentación con retraso motor y debilidad con sobrevivencia hasta la edad adulta. Regula sistemas de degradación y remplazo proteico.
- LMOD3: se han asociado sobre todo a formas neonatales graves aunque hay casos descritos con formas típicas. Participa en la contracción muscular.
- YOQ18B: descrita durante el 2018.
- MYPN: inicio en primer década, lentamente progresiva, atrofia debilidad muscular cervical y proximal en miembros inferiores, posible afectación leve respiratorio y/o cardíaca. Participa en la contracción muscular. Descrita durante el 2018.
- TNNT3: participa en la contracción muscular.
- GYG1 y RYR3: recientemente se han reportado mutaciones en otros dos genes, como posible causa de MN, pero deben realizarse más estudios para confirmarlo.
En la actualidad, aún hay personas con MN sin diagnóstico molecular específico.
¿Cómo se transmite?
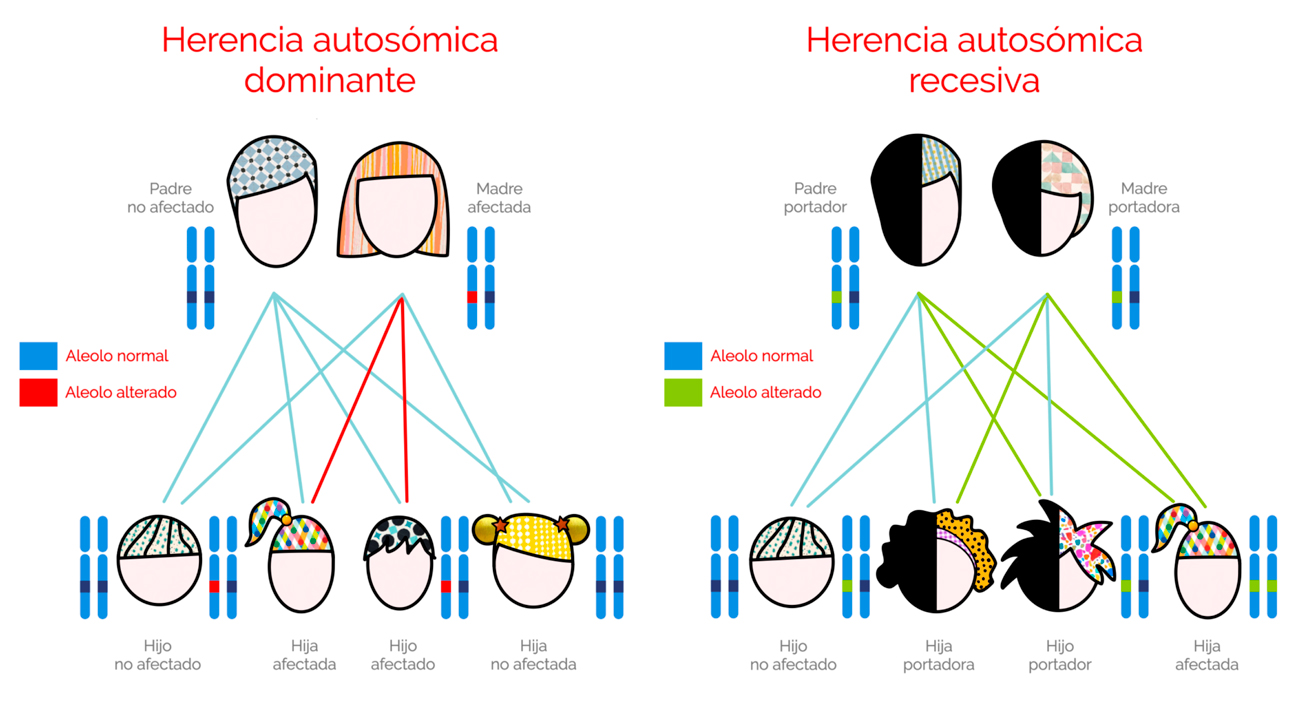
Cada persona hereda dos alelos (copias) del mismo gen: el materno y el paterno. Según estos sean, la MN puede ser heredada de forma:
- Autosómica Dominante: el alelo alterado es dominante sobre el normal bastando una sola copia para que se exprese la enfermedad. Al ser autosómico, el gen se encuentra en uno de los 22 pares de cromosomas no sexuales, o autosomas, pudiendo afectar con igual probabilidad a hijos e hijas. El alelo alterado puede haber sido heredado tanto del padre como de la madre. Normalmente se manifiesta en todas las generaciones de una misma familia. Cada persona afectada tiene normalmente un progenitor afectado y una probabilidad del 50% de que un hijo o hija nazca con la patología. Estos posibles resultados suceden al azar, manteniéndose la probabilidad de igual manera en cada embarazo y entre hijos e hijas.
- Autosómica Recesiva: el alelo alterado al ser autosómico, también se encuentra en uno de los 22 pares de cromosomas no sexuales pudiendo afectar por tanto con igual probabilidad a hijos e hijas. Sin embargo, son necesarias dos copias del gen para que se exprese la enfermedad. Por esta razón, las copias del gen alterado deben estar presentes tanto en el padre como en la madre. En este tipo de herencia, la patología no suele manifestarse en todas las generaciones, pudiendo saltarse una de ellas apareciendo por ejemplo en abuelos y nietos.
- De Novo: en ocasiones la persona con la enfermedad genética dominante es la primera afectada en la familia. Esto sucede cuando es una nueva mutación del gen producida durante el periodo de formación del óvulo o el espermatozoide. Cuando esto sucede ni el padre ni la madre portan la enfermedad, siendo muy poco probable que otro hijo o hija la tenga, aunque siempre es recomendable el consejo genético. Sin embargo, el niño o niña con la enfermedad, tiene el gen mutado por lo que puede transmitirlo a su descendencia.
Una mutación genética, en la actualidad, no puede ser corregida por lo que está presente durante toda la vida.
¿Es posible el diagnóstico prenatal?
El diagnóstico prenatal es factible siempre y cuando se realice previamente el estudio genético que identifique la mutación familiar causante.
¿Cómo se diagnostica?
La MN se diagnostica a través de:
- Manifestaciones clínicas y exploración física.
- Biopsia muscular con hallazgos de cuerpos nemalínicos.
- Estudio neurofisiológico.
- Resonancia magnética muscular de cuerpo entero.
- Estudio genético.
Asimismo, el diagnóstico genético se ve dificultado por:
- Número elevado de genes responsables.
- Baja incidencia.
- Heterogeneidad fenotipíca-genotípica (convergencia fenotípica-divergencia genotípica).
- Número, tamaño y complejidad de algunos genes.
¿Qué son los cuerpos nemalínicos?
Los cuerpos nemalínicos son estructuras en forma de bastoncitos compuestas por acúmulos de proteínas. Pueden detectarse en otras patologías como otras miopatías hereditarias o miopatías mitocondriales, inflamatorias e infecciosas. Considerándose que la formación de cuerpos nemalínicos representa una respuesta del músculo a determinadas situaciones patológicas.
Su formación, se cree debida a un mal plegamiento de la proteína mutada, lo cual la lleva a no poder realizar bien su función.
Hoy, la duda reside en si los cuerpos nemalínicos empeoran la MN o únicamente tienen un carácter secundario dentro de la misma.
¿Cómo se aborda?
El abordaje de la MN debe recaer en un equipo multidisciplinar integrado por especialistas en enfermedades neuromusculares.
Al no existir una terapia curativa, el objetivo es de carácter paliativo y dirigido a evitar, retrasar y disminuir las manifestaciones de manera específica en cada caso con el fin de lograr el máximo nivel funcional y de independencia posible.
Dicho abordaje pretende:
- Mantener la fuerza.
- Mantener la movilidad (prevenir contracturas articulares).
- Controlar las complicaciones ortopédicas (contracturas y escoliosis)
- Evitar y eliminar el dolor.
- Conservar la función respiratoria.
- Conservar la función cardíaca.
- Conservar la densidad ósea.
- Lograr y mantener un buen estado nutricional, un óptimo estado de hidratación y un control (en periodos de ayuno y de estrés por infecciones) de la propensión a sufrir hipoglucemias.
- Minimizar los problemas de deglución.
- Conseguir un lenguaje comprensible.
- Alcanzar el máximo grado de independencia.
- Reducir las hospitalizaciones.
- Mantener un buen nivel de autoestima.
Los medios empleados serán:
- Fisioterapia: ejercida ininterrumpidamente con el fin de mantener la fuerza muscular, el rango de movimiento, prevenir la escoliosis, minimizar o evitar el dolor y conservar y mejorar la movilidad así como el grado de independencia en las actividades cotidianas.
- Fisioterapia respiratoria y asistencia a la tos cuando se presenta debilidad de la musculatura respiratoria. Los cuidadores deben recibir entrenamiento en cuanto a resucitación cardiopulmonar y otras técnicas de rescate, como aquellas para el ahogo.
- Soporte respiratorio: control respiratorio por neumología, resultando necesario, en un gran número de casos, soporte respiratorio que incluye oxigenoterapia y o ventilación mecánica no invasiva nocturna. Así mismo, las infecciones respiratorias deben ser tratadas de manera precoz e intensiva. En los casos más severos resulta vital la traqueotomía.
- Estudio de alteraciones de la deglución (disfagia) y malnutrición: estando indicado el trabajo de logopedia así como fórmulas hipercalóricas, alimentación por sonda nasogástrica o sonda de gastrostomía (botón gástrico)…
- Estudio y control de osteoporosis, osteopenia y déficit de calcio y vitamina D.
- Logopedia: en la mayoría de los casos debido a problemas en la articulación del lenguaje y por problemas de deglución.
- Terapia ocupacional: imprescindible para mantener y alcanzar el mayor grado de independencia en la vida diaria, facilitando la adaptación a la discapacidad provocada.
- Control médico por rehabilitador conjuntamente con traumatólogo: control y revisión de problemas articulares y vertebrales con el fin de detectar precozmente las contracturas o la escoliosis. Suelen realizarse intervenciones tempranas para prevenir cambios permanentes en las extremidades (deformidades) o complicaciones respiratorias en el futuro. Además se estudian y prescriben los dispositivos necesarios para aumentar o mantener la independencia de la persona.
- Control cardiológico: aún siendo raras las complicaciones cardíacas, todas las personas afectadas por MN deben estar sometidas a revisión cardiológica.
- Control oftalmopléjico: es común que el párpado no llegue a cerrarse del todo durante el sueño pudiendo provocar que la capa externa de la cornea se raye. Una cornea rayada es dolorosa y puede causar la disminución de la visión. La debilidad de los músculos oculares también puede hacer difícil la tarea de mirar hacia muchas direcciones sin mover la cabeza (oftalmoplejía).
- Control del dolor muscular y la fatiga: la causa exacta del dolor muscular es desconocida, pero puede ser debida por la propia enfermedad, por retracción articular, huesos más delgados o deformaciones en las articulaciones que se desarrollan con el tiempo. La fatiga es producto de la debilidad muscular de base.
- Consejo genético: debe realizarse tanto para las personas afectadas como para sus familias.
Un punto aparte merecen las diversas y heterogéneas estrategias terapéuticas empleadas. Dada la baja prevalencia de la MN, su gran variabilidad, la falta de investigación así como la desconexión y diversidad de opiniones de quien atiende cada caso, las estrategias terapéuticas son escasas y poco empleadas pese a las mejoras encontradas con su toma en casos publicados. Así nos encontramos con el uso de:
- Piridogstigmina (Mestinon): La piridostigmina es un inhibidor reversible de la colinesterasa, enzima encargada del metabolismo e inactivación de la acetilcolina. “Después de 3 días se observó una respuesta beneficiosa dramática, con una notable mejora en la ventilación. Después de 2 semanas se había observado una significativa mejoría clínica objetiva: movimientos espontáneos se incrementaron notablemente y su cara mostró más expresión. El control de la cabeza fue adquirido después de 4 semanas de tratamiento (Fig. 2), después de 2 meses estaba sentada sin ayuda.” (Publicación).
- L-Carnitina: Su principal función es la de generar energía para nuestro organismo. Es un elemento clave para la correcta oxidación de los ácidos grasos en la mitocondria, y así liberar energía en forma de ATP (adenosín trifosfato).
- L-Tirosina: aminoácido no esencial derivado en condiciones normales del aminoácido esencial fenilalanina, incluyendo los requerimientos totales diarios de éste último a los de la tirosina. La L-tirosina se encuentra no obstante ampliamente distribuida en las proteínas y enzimas corporales, poseyendo numerosos roles funcionales, entre los que destacan la síntesis de neurotransmisores involucrados en la regulación de la coordinación motora, el comportamiento, el aprendizaje, la memoria, la regulación de los niveles de humor y la neutralización de radicales libres. (Publicaciones)
- Coenzima Q10: compuesto que el cuerpo elabora de manera natural, empleándolo para el crecimiento celular y como antioxidante (protege a las células de las sustancias químicas, radicales libres)
- Salbutamol: broncodilatador que relaja los músculos de las paredes de las pequeñas vías aéreas de los pulmones. Abre las vías respiratorias aliviando la opresión en el pecho, las sibilancias y la tos. Ayuda a prevenir la falta de aire y las sibilancias durante el ejercicio.
Posibles futuras opciones terapéuticas en base a lo que se sabe de la MN son:
- Tratamientos capaces de modular los mecanismos de la excitación-contracción muscular así como el manejo del calcio intracelular, los cuales se ha demostrado son unas de las principales alteraciones en el mal funcionamiento muscular en la MN.
- Terapias que modulen el sistema ubiquitina-proteasoma ya que en el músculo esquelético de pacientes con MN se ha observado una disminución de los componentes de dicho sistema, lo que sugiere la existencia de una alteración en el recambio proteico muscular.
- Fármacos capaces de modificar la autofagia (mecanismo natural de regeneración que ocurre en nuestro cuerpo a nivel celular que se ha detectado alterada en las células musculares en la MN.
- Plegamiento de proteínas: Las chaperonas (conjunto de proteínas presentes en todas las células, muchas de las cuales son proteínas de choque término, y cuya función es la de ayudar al plegamiento de otras proteínas recién formadas en la síntesis de proteínas) son esenciales para el ensamblaje del sarcómero y el correcto funcionamiento de las proteínas musculares.
- La regulación del gen de la actina cardíaca en el músculo esquelético en etapa postnatal, consiguiendo mejorar la fuerza y la actividad motora.
- Modificación epigenética para combatir la debilidad muscular.
¿Cuál es su evolución?
La MN puede presentarse como grave, moderada o leve siendo su evolución, por lo general, estática o muy lentamente progresiva.
Las complicaciones clínicas graves son casi siempre secundarias a la deficiencia respiratoria y en ocasiones a problemas cardiológicos. No obstante puede darse la combinación de MN y cardiopatía, aunque es poco frecuente. En la mayoría de los casos la cardiomiopatía se desarrolla en la edad adulta, mientras que en escasas ocasiones lo hace en la infancia.
En la presentación moderada y leve, la expectativa de vida es normal pese a verse considerablemente mermada la calidad de la misma (especialmente en la presentación moderada).
Momentos cruciales como la pubertad, deben ser especialmente vigilados debido a que la musculatura toma más tiempo en adaptarse al crecimiento del esqueleto afectando esto directamente a la marcha y agravando los problemas de control posicional los cuales derivan en problemas con el sistema esquelético.
Ya en la edad adulta deben vigilarse además los siguientes aspectos:
- Aumento de la debilidad debido al aumento de tamaño y peso, así como por el natural paso de los años.
- Posible desarrollo de un nuevo trastorno independiente de la MN.
- Enfermedades como diabetes, obesidad, ataques cardíacos y ACV (más prevalentes en las personas a medida que envejecen).
- Planificación familiar.
Miopatía Nemalínica esporádica de inicio tardío (SLONM)
La miopatía nemalínica esporádica de inicio tardío (SLONM) es una miopatía adquirida poco frecuente (>1/1.000.000 de nacidos vivos), reconocida por primera vez en 1966.
Se presenta, normalmente a partir de los cuarenta años (aunque existen casos mucho más tempranos), con debilidad predominantemente proximal de las extremidades superiores y / o inferiores así como debilidad de los extensores del cuello, disnea y el hallazgo de cuerpos nemalínicos en la biopsia muscular.
De igual manera puede presentar:
- Insuficiencia respiratoria, desde el inicio y normalmente severa, requiriéndose soporte ventilatorio.
- Afectación cardíaca de importancia.
- Atrofia muscular severa.
- Problemas deglutorios, requiriéndose alimentación por gastrostomía.
- Debilidad facial.
- Disartría (Dificultad para articular sonidos y palabras causada por una parálisis o una ataxia de los centros nerviosos que rigen los órganos fonatorios.)
- Mialgia (dolor muscular).
- Fasciculaciones (pequeñas e involuntarias contracciones musculares, visibles bajo la piel y que no producen movimiento de miembros, debidas a descargas nerviosas espontáneas en grupos de fibras musculares esqueléticas.)
- Oftalmoparesis (incapacidad para mover voluntariamente el globo ocular. Es la parálisis de uno o más músculos oculares).
Pudiendo también asociarse con otros cambios menos específicos como:
- Aferencias atróficas.
- Células lobuladas.
- Anomalías mitocondriales.
- El músculo cardíaco estriado puede ser susceptible al mismo mecanismo dañino que afecta al músculo esquelético (debe confirmarse mediante estudios de patología).
Diagnóstico diferencial:
- Miositis esporádica del cuerpo de inclusión (sIBM).
- Miopatías inflamatorias idiopáticas (IIM).
- Miopatía por VIH.
- Miopatía por sífilis.
- Esclerosis lateral amiotrófica (ELA).
Pruebas diagnósticas:
- Biopsia muscular con presencia de cuerpos nemalínicos.
- Exámenes de laboratorio: muestran un CPK normal o ligeramente elevada.
- Pruebas negativas de anticuerpos anti-VIH y sífilis.
- Inmunoelectroforesis: puede revelar una gammapatía monoclonal de significación incierta en muchas de las personas afectadas, lo cual parece complicar el pronóstico.
- Biopsia de médula ósea: puede mostrar infiltración intersticial de células plasmáticas monoclonales.
- Aspiración de médula ósea: puede revelar una célula plasmática anormal en filtración.
- Pruebas respiratorias.
- Electromiografía con aguja: revela potenciales de unidades motoras miopáticas, ocasionalmente asociados con potenciales de bridación en reposo.
- Gammagrafía orofaríngeo-sophageal: evaluación de la deglución.
- IRM del músculo de la cintura superior e inferior.
- Resonancia magnética muscular: sale alterada, más los datos sobre imágenes musculares, útiles como herramienta de diagnóstico en miopatías genéticas y adquiridas aún faltan para esta en particular.
- Estudio cardiológico: monitorización ECG de 24 h; ecocardiograma; resonancia magnética cardíaca y angiografía coronaria diagnóstica.
La enfermedad se asocia generalmente con un pronóstico muy precario debido principalmente a la afectación respiratoria. Dicho pronóstico parece ser peor si se encuentra asociada la MN a una gammapatía IgG monoclonal.Los últimos estudios sugieren un espectro más amplio de participación, que incluye casos atípicos.
El diagnóstico temprano (prácticamente inexistente por el desconocimiento y rareza de la patología) y el tratamiento parecen ser factores muy importantes para una posible recuperación.
Posibles terapias farmacológicas son:
- Esteroides orales e intravenosos.
- Ciclos repetidos de inmunoglobulinas intravenosas mensuales.
- Melfalán.
- Bortezomib(Velcade).
- Tratamiento con melfalán seguido de autotrasplante de células madre (aSCT) ha dado resultados alentadores en SLONM-MGUS (gammapatía monoclonal de significación desconocida).
- Inmunomoduladores e inmunosupresores.
- Plasmaféresis (método mediante el cual se extrae completamente la sangre del cuerpo y se procesa de forma que los glóbulos blancos, glóbulos rojos y plaquetas se separen del plasma. Las células de la sangre se devuelven luego al paciente sin el plasma, el cual el organismo sustituye de inmediato).
El tratamiento precoz con corticoesteroides, inmunosupresores y / o IgIV puede tener un impacto favorable en la progresión clínica de pacientes sospechosos de SLONM y un TCS como «terapia de rescate» puede reservarse para no respondedores con SLONM-MGUS confirmado.
La falta de un diagnóstico temprano (prácticamente inexistente por el desconocimiento y rareza de la patología) es uno de los principales enemigos del SLONM, una miopatía grave pero potencialmente tratable en la mayoría de los casos si es precozmente detectada, estudiada y tratada en su conjunto.
